

转自:药渡
2023年9月,第二届中国生物计算大会已于苏州盛大落幕,但聚焦于AI/计算的浪潮从不曾停歇。以新一代人工智能分子模拟算法体系为核心特色的新兴计算企业予路乾行逐渐走入了大众视野,本篇将详细介绍予路乾行的平台亮点、应用场景和案例分享。
“10年、10亿美金、10%的成功率”常被认为是医药研发行业难以打破的“魔咒”。1981年,《财富》杂志宣称计算机辅助的药物研发是“下一次工业革命”。随着时代的变革,在人工智能快速发展的今天,予路乾行开发并融合了基于多尺度分子动力学、人工智能和量子力学的新一代人工智能分子模拟算法体系,通过从分子层面揭示生物大分子柔性机制,引领新药研发从分子照片走入分子电影时代,能够将传统新药研发PCC前上千次化合物合成试错成本有效降低至百次。

1
Divamics平台介绍
予路乾行基于多尺度分子动力学结合自主创新的AI算法而构建了新一代人工智能药物研发平台Divamics,提供各类计算化学服务,包括但不限于高柔性靶点建模、靶点可成药性分析、面向大分子及小分子药物结构筛选、设计、优化等,特别是针对难成药靶点提供分子成药机理解析、全新药物分子结构设计、和结构优化解决方案。Divamics平台核心特点在于寻找运算精度和效率之间的最佳平衡点。在明确靶点的构象特征与结合位点的同时,小分子药物与靶标蛋白结合后如何改变其构象特征也是分子药效学的核心问题。
予路乾行采用多尺度分子动力学对于生理环境下的生物大分子的构象特征和运动模式进行全方位模拟,从第一性原理出发,结合量子力学、牛顿力学和机器学习开发的分子力场有效地提升了力场精度,同时,基于蒙特卡洛法开发的平行采样算法和引入了高斯动力学、元动力学、交换副本动力学等增强采样技术大大提高了运算的效率,有效地平衡了运算精度与效率。通过热力学与动力学角度的双重评估,为新药研发提供更全面的分子层面机理信息。
2
应用场景
生物大分子体系动态结构建模
1
蛋白质三维结构的从头/同源建模和优化
靶点-配体复合物结构建模,以及靶点、配体分子各自在自由态/结合态的构象特征分析
蛋白质-蛋白质结合界面(PPI)建模与重要残基分析
跨膜体系(生物膜、跨膜蛋白等生物大分子)的构建和优化
固有无序蛋白(IDP)优势构象群的构建
难成药靶点机制探索
2
难成药靶点蛋白建模和优化
难成药靶点蛋白新结合位点的探索和苗头化合物发现
难成药靶点蛋白与药物分子的结合/解离路径探索和机理解释
靶点蛋白别构结合位点发现与分子药效学机理探索
多聚体蛋白质形成过程的结合/解离路径探索和机理解释
固有无序蛋白(IDP)与药物分子相互诱导形成的优势结合构象群的构建
动力学虚拟筛选
3
基于符合Lipinski/Pfizer /Golden Triangle Rules标准的类药化合物分子结构初筛
基于分子动力学的靶点多构象筛选
基于药效团模型及诱导契合对接、打分的虚拟筛选运算流程
基于AI力场驱动的多尺度分子动力学模拟及结合自由能分析
小分子药物设计和结构优化
4
提升先导化合物的蛋白与细胞活性(Kd, IC50等)
提高药物靶向选择性,降低脱靶效应
针对别构位点结合的药物结构优化
针对结合蛋白质多聚体的药物结构优化
针对固有无序蛋白(IDP)的药物结构优化
共价结合药物分子的靶向选择性优化及弹头优化
药物代谢位点识别与药物相互作用机理探索
PROTAC药物设计和结构优化
5
泛素连接酶的选择
靶向POI的弹头设计和优化
linker的筛选和优化
3
案例分享
1
准确预测药物-靶点的结合模式
Fig1.1抑制剂与靶点结合过程模拟

Fig1.2抑制剂与靶点结合/解离路径探索

Fig1.3模拟生成的最优结合模式对比实验获得的共晶结构

该项目需求为预测靶点和抑制剂分子的结合模式,探索化合物的药效学分子机制,及其对靶点相关结构域的构象诱导改变能力。予路乾行利用分子动力学平台中的“核武器”——元动力学增强采样技术,对抑制剂分子与靶点的结合与解离过程进行全程模拟(Fig1.1),得到了准确的配体-靶点优势结合模式,与客户通过实验解析得到的X射线晶体衍射结构进行对比,误差(RMSD)小于0.5埃(Fig1.3);同时通过对药物-靶点相互作用的机制探索,在分子层面上阐释了由于配体结合诱导靶点构象发生改变,使得配体与靶点发生结合和解离时倾向于采取不同路径的原因(Fig1.2)。
2
PPI建模与靶点成药机制探索
Fig2.1 自由能模拟及分子动力学模拟图
路径A分子动力学模拟

路径B分子动力学模拟


Fig2.2结合界面处能量示意图

Fig2.3抑制剂(紫色)破坏蛋白-蛋白结合过程的分子动力学模拟图

该靶点为中枢神经系统疾病靶点,位于复杂信号通路中,且结合位点与作用机制不明。通过动力学模拟,予路乾行通过建模判断最优的小分子药物位点为蛋白-蛋白结合界面(interrupt protein-proteininteraction),通过设计小分子抑制剂通过进入结合界面后,破坏蛋白-蛋白间关键的盐桥,从而可以达到阻止上下游蛋白结合的作用。依此分子机制,予路乾行团队筛选了上亿数量级的类药化合物库,2周内交付6个化合物结构,首批交付化合物细胞活性实验结果IC50达1-10uM。目前正在支持合作方完成化合物专利的申报。
同时,由于该药物的研发存在透过血脑屏障需求,需达到一定的脑内分布浓度。予路乾行搭建了一套生物膜模拟平台,通过模拟溶液中化合物穿透细胞膜并扩散的平均速率与自由能变化评估药物透过血脑屏障的能力。同通过预测药物与脑内相关蛋白质的非特异性结合能力给予药物脑内生物利用度的综合运算评估。目前结合药物透血脑屏障运算分析与动物实验验证,目前予路乾行所设计的最优化合物在小鼠脑内的分布浓度相较阳参化合物提高了18倍左右。
Fig2.4化合物团簇与细胞膜融合的过程

备注:黄色球状小分子为外源性化合物,下层为双层细胞膜,蓝色的为POPC,绿色的为POPS。
予路乾行自主研发的生物膜构建和膜体系分子力场能够精确模拟化合物团簇与不同组分细胞膜的融合过程(Fig2.4),对药物分子的分布、吸收、代谢和外排机理提供全方位信息。
Fig2.5两种不同化合物分子在生物膜上的扩散速率对比


予路乾行通过在自主研发的膜体系环境中进行分子动力学模拟,可获得不同备选化合物分子(Fig2.5中展示两种)在生物膜上的扩散速率,从而定量描述不同化合物分子透膜能力的差异。
3
提升PROTAC降解活性
Fig3两个结构非常相似的PROTAC三元复合物体系
Fig3.1没有降解活性的PROTAC分子动力学模拟图
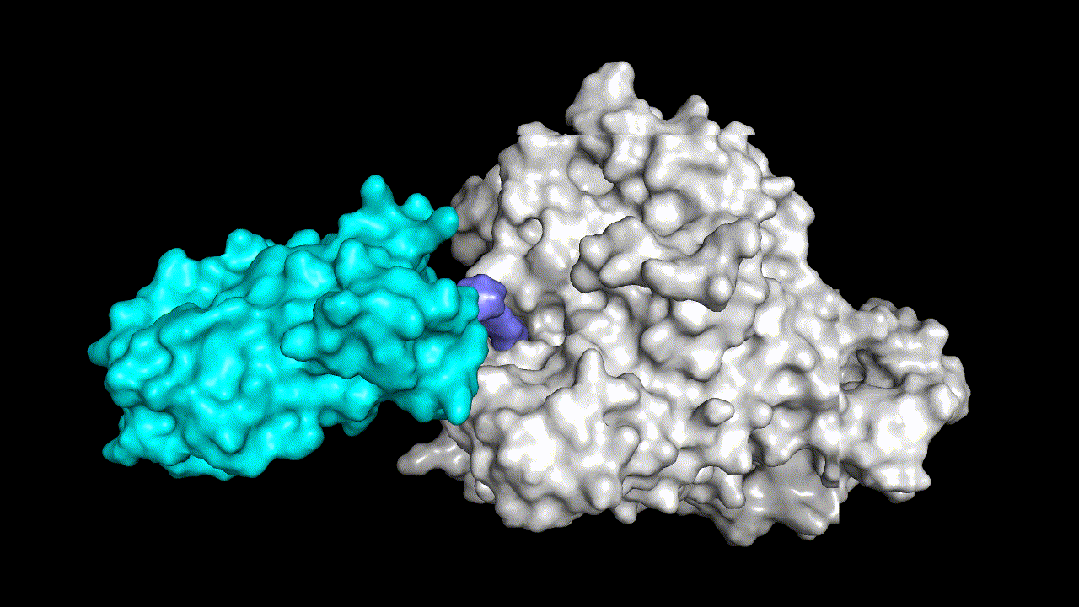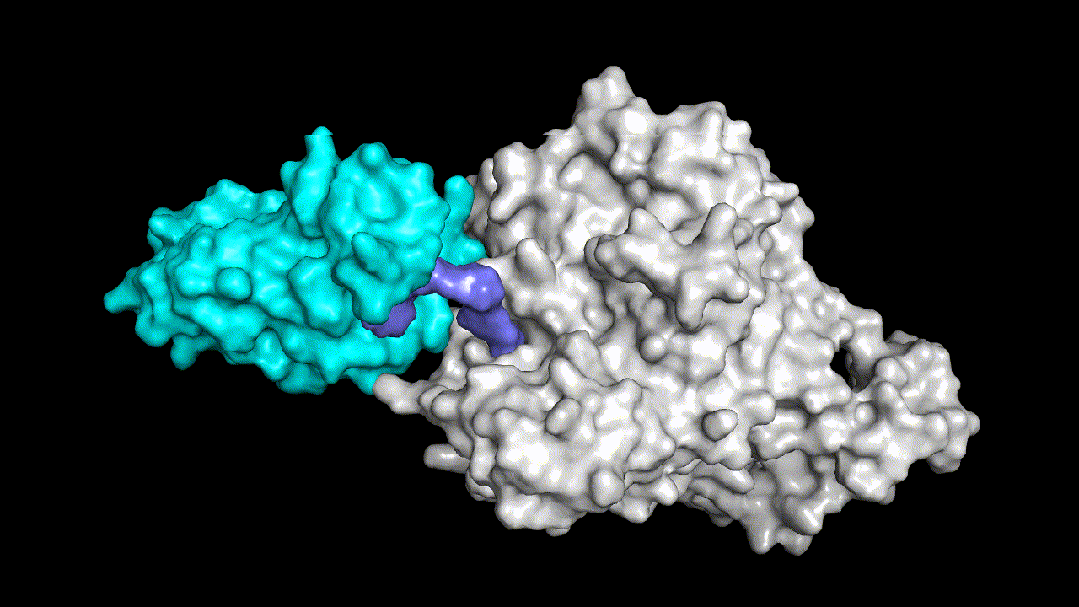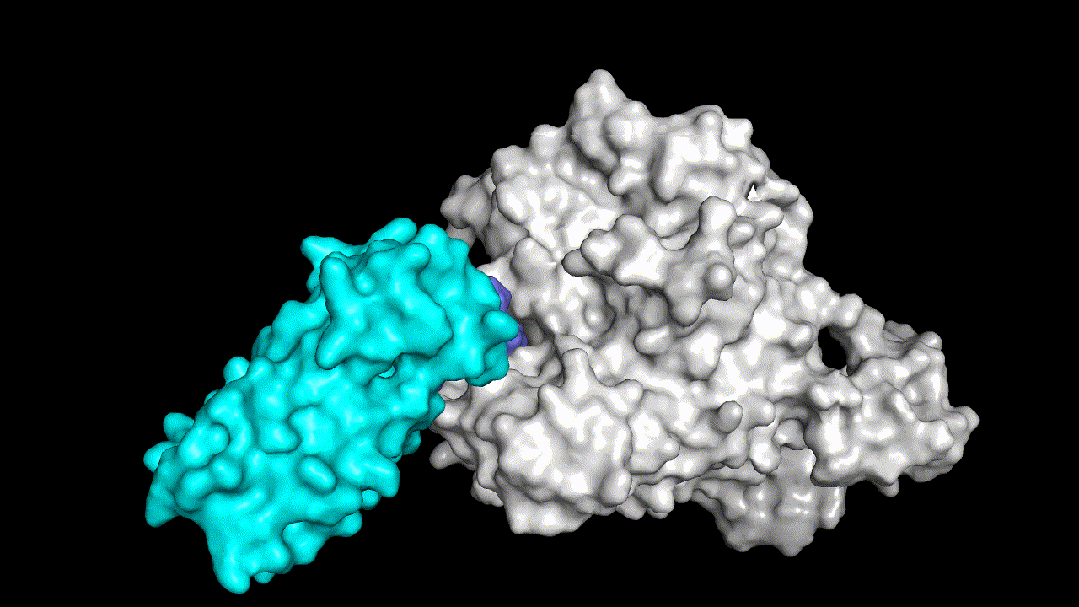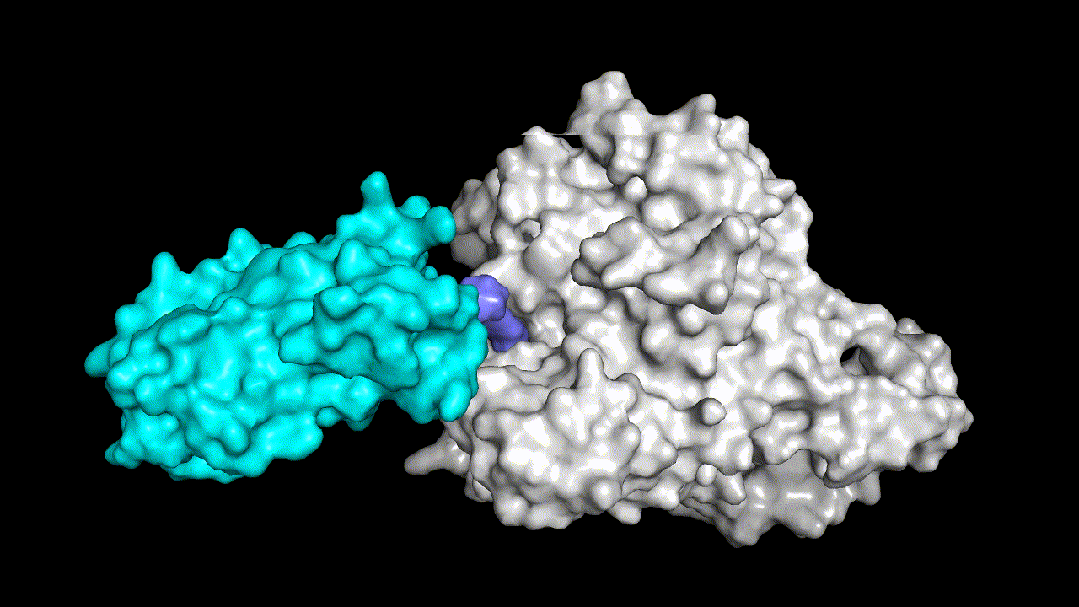
Fig3.2有降解活性的PROTAC分子动力学模拟图
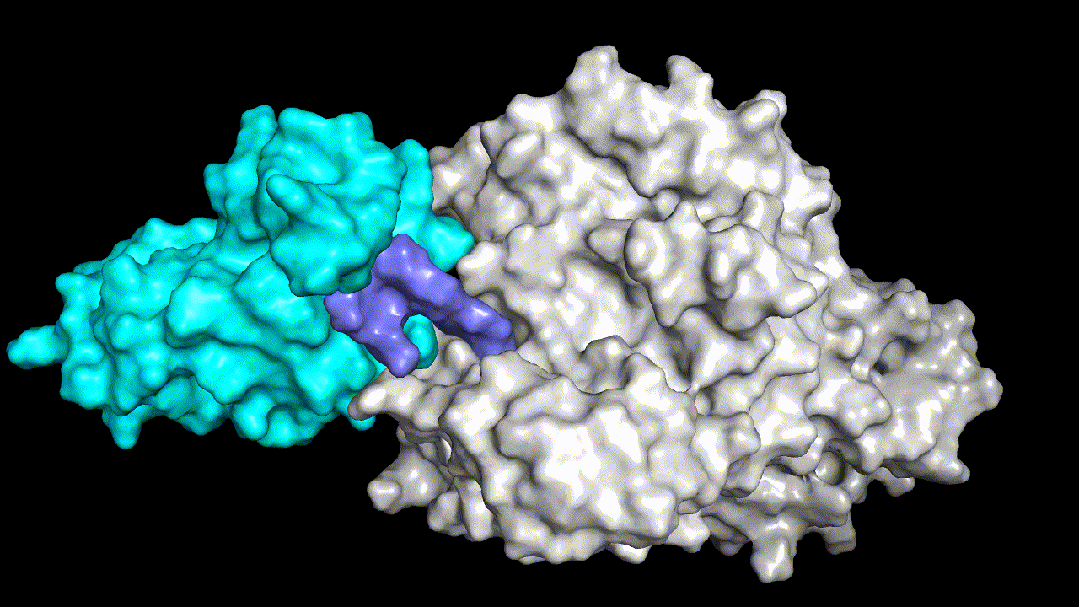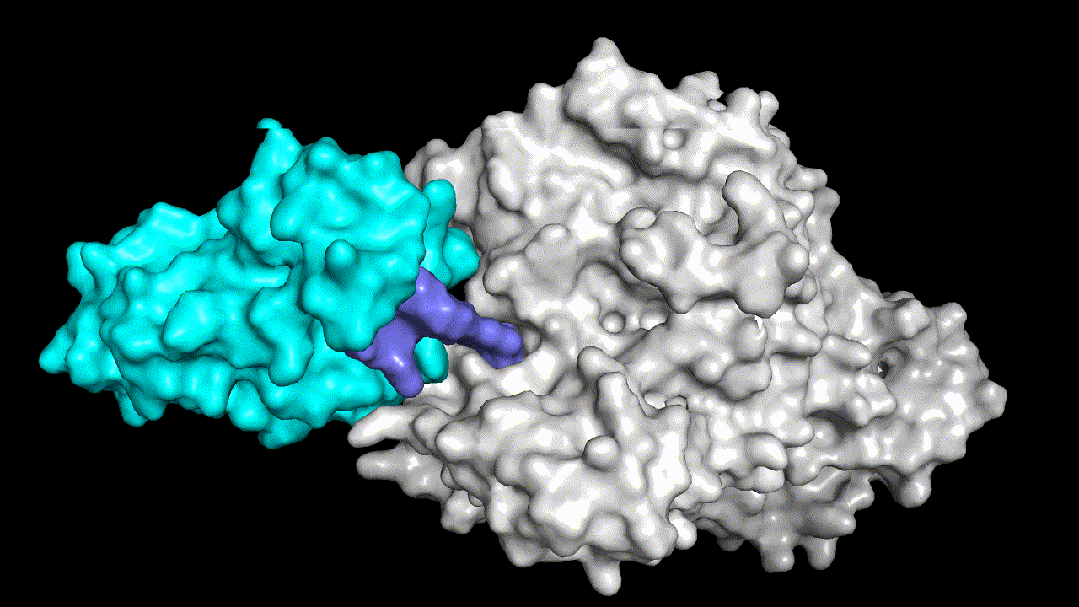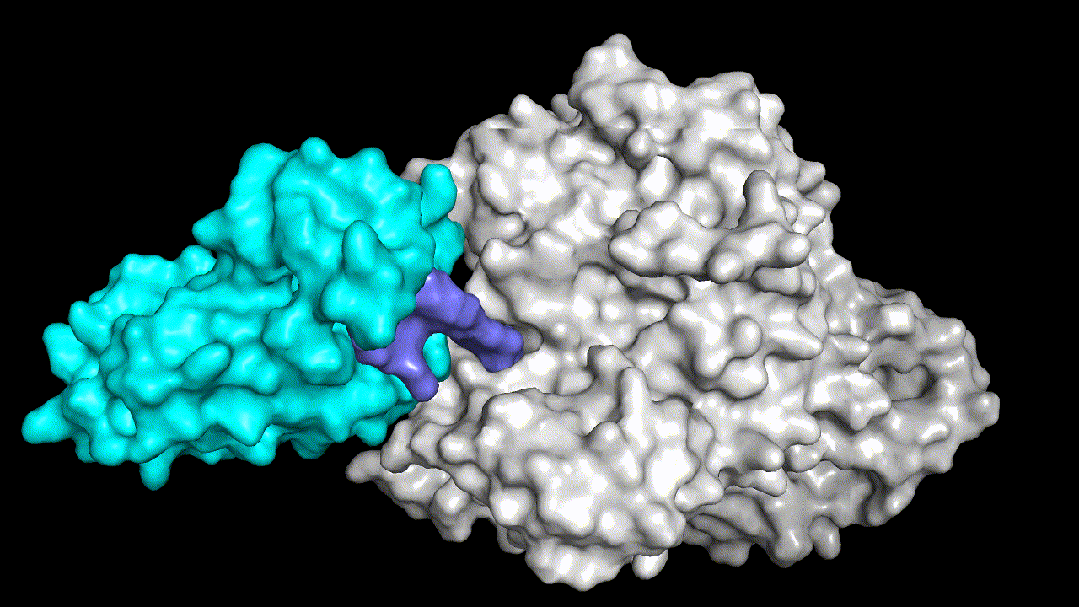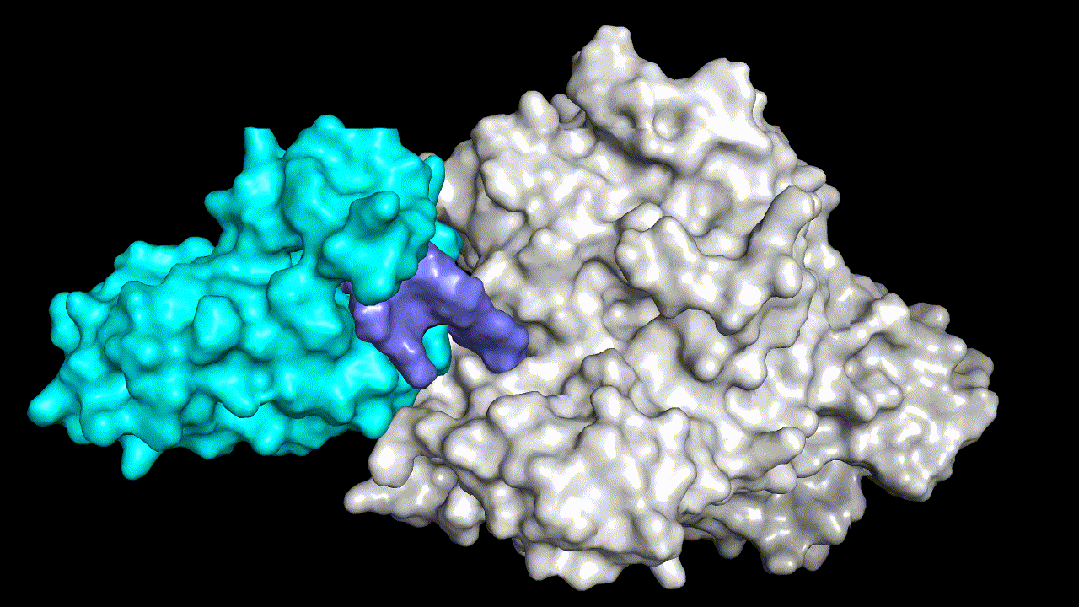
Fig3.3予路乾行特色PROTAC药物设计平台

予路乾行基于核心算法和多尺度分子动力学模拟技术,构建了能够覆盖warhead-目标蛋白结合模式的确定和优化、泛素连接酶及其binder的选择和优化、linker的筛选及优化等诸多PROTAC药物分子设计全流程中场景的AIDD平台。Fig3展示了予路乾行通过结构优化提升PROTAC降解活性的案例,Fig3.1中所示的PROTAC分子没有降解活性,需要找到导致该现象的原因,明确其对应的分子机制。予路乾行通过对该三元复合物体系进行构建并进行动力学模拟,发现该体系的linker结构导致了体系柔性的提升,从而无法在目标蛋白和泛素连接酶之间形成稳定的PPI,进而造成体系稳定性和降解活性的显著下降。锚定PPI界面上的两处关键盐桥氢键,予路乾行通过优化linker结构,使两处盐桥氢键的生成概率分别由原来的77.3%和0%提升至98.5%和94.4%,使得优化后的PROTAC分子所在的三元复合物体系(Fig3.2所示)获得了稳定的PPI界面和降解活性。予路乾行打造的基于多尺度深度学习的人工智能PROTAC分子设计平台不仅可以优化PROTAC结合稳定性,提升降解活性,还能有效降低脱靶效应和钩子效应对结合模式和结合强度的影响。
4
结语
借助基于多尺度深度学习和分子动力学模拟技术的药物分子设计平台,予路乾行得以从分子层面揭示靶点柔性机制用以指导药物分子设计,帮助新药研发企业解决药研早期的各项难题,引领新药研发从分子照片走入分子电影时代。得名于《周易》“终日乾乾,与时偕行”的予路乾行,将如其中所言,深耕技术,自强不息,一路前行,永不休止。
关于予路乾行
予路乾行是一家以人工智能、量子力学及分子模拟算法为核心技术,通过模拟运算赋能新药研发的交叉学科技术驱动型新药研发CRO公司。予路乾行开发并融合了基于多尺度分子动力学、人工智能和量子力学的新一代人工智能分子模拟算法体系,通过从分子层面揭示生物大分子柔性机制,引领新药研发从分子照片时代进入分子电影时代,为新药研发早期阶段的诸多难题提供解决方案,将传统新药研发PCC前上千次化合物合成式错成本有效降低至百次。
公司于2011年底创立之初即获评苏州市工业园区科技领军项目企业,次年获批苏州市级姑苏领军人才项目,核心算法获得诺贝尔化学奖得主Roald Hoffmann推荐,目前已与多家国内外生物医药企业合作推进数条新药研发管线并取得多个突破性成果,达成近20个项目里程碑。
