

转自:药时空
T细胞通过T细胞受体(TCR)识别MHC蛋白上的短肽抗原,以抗原特异性的方式对威胁做出反应。T细胞与其靶细胞之间的TCR-肽-MHC相互作用决定其功能,从而影响其在疾病中的作用。缺乏发现抗原的方法限制了对整个T细胞反应的抗原图景的基本了解。高通量测序、质谱细胞术、微流体学和计算生物学的最新进展导致了应对T细胞抗原发现挑战的方法激增。
T细胞抗原发现的挑战
T细胞在清除病原体和监测病变细胞中起着至关重要的作用。它们具有通过表面TCR区分自身和非自身抗原的能力。大多数人类T细胞表达经典的TCRs,由TCRα链和TCRβ链组成,与CD3链(γ,δ,ε和ζ)共表达(图1a)。TCRα和TCRβ链分别由可变区域(V)和恒定区域(C)组成。Vα链由V和连接(J)基因片段(TRAV和TRAJ)编码,而Vβ链由V、多样基因(D)和J基因片段(TRBV、TRBD和TRBJ)组成。每个基因片段的多个拷贝在基因组中编码,并按V-J或V-D-J顺序组合重排,以分别在TCRα和TCRβ链中产生多样性(图1b)。额外的多样性是通过基因连接边界上的非模板化核苷酸(N)添加和/或删除来产生的。位于TCR V结构域的三个互补决定区(CDR)是其与多肽-MHC(pMHC)复合体相互作用的主要原因,从而赋予TCR抗原特异性。高通量测序研究的结果表明,TCR重复序列的实际多样性可能在10^11-10^12个独特的TCRs的范围内。当TCR识别同源的pMHC复合体时,CD3链上的基于免疫受体酪氨酸的激活基序(ITAMs)立即被磷酸化,触发信号级联,导致T细胞激活和诱导T细胞功能。
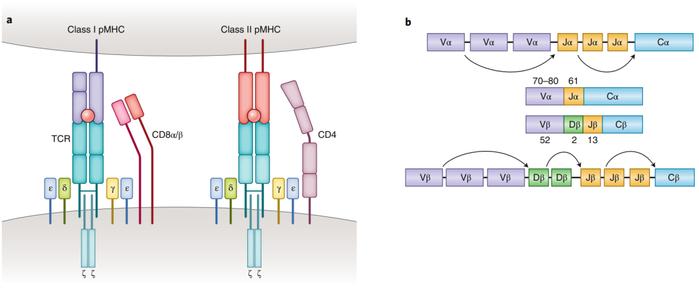
●图1 TCR-pMHC识别的分子基础。经典的T细胞受体αβ复合体含有CD3ζ、CD3γ、CD3δ和CD3ε链。MHC I分子由α链和β2-微球蛋白链组成。MHC II分子由两条非共价结合的α和β链组成。α和β链的遗传重组是通过体细胞V(D)J重组而产生的。
TCR-pMHC相互作用是获得性免疫的关键因素。经典的CD8+和CD4+αβΤ细胞分别识别MHC I类和II类分子呈递的短肽。与这些MHC限制性αβT细胞形成鲜明对比的是,由TCRγ和TCRδ链组成的非常规γδT细胞可以不受MHC限制地识别非肽类抗原(如脂类抗原)。多肽表位可以通过胞浆途径(MHC-I限制性表位)或胞内途径(MHC-II限制性表位)展示在细胞表面。TCR-pMHC相互作用的亲和力通过辅助受体CD4和CD8分别与MHC II和MHC I结合而增强。TCR-pMHC相互作用的另一个特征是混杂:每个TCR能够识别数百万不同的多肽,而许多不同的TCR可以识别给定的多肽。然而,技术限制阻碍了对TCR-pMHC相互作用图景的广泛绘制。能够可靠识别TCR配体的方法将解决这一未得到满足的需求,并极大地帮助自身免疫、传染病和癌症免疫学领域。
理解控制TCR-pMHC相互作用的分子规则的挑战由四个主要因素复杂化。首先,针对特定抗原的T细胞在外周血液中的出现频率非常低,使得准确检测这些罕见的抗原特异性群体变得困难。其次,TCR和pMHC都是多特异性的,允许以最小的序列相似性识别多肽。第三,TCR-pMHC相互作用的亲和力比其配体抗体的亲和力低几个数量级,需要灵敏的生化技术进行检测。第四,抗原的加工和提呈产生了大量潜在的T细胞表位,使得在抗原提呈细胞(APC)中合成或表达它们以进行筛选变得困难。已经开发了几种计算工具,如NetMHC,来预测MHC结合,但这些工具仍然不能准确地预测这些多肽的免疫原性。
以抗原为导向的方法
已经开发了各种抗原导向的方法来表征抗原特异的T细胞(图2)。经典的策略是通过评估T细胞在暴露于特定抗原后的功能反应来检测TCR-pMHC的相互作用。这些方法包括T细胞增殖的测量、用铬释放法检测T细胞的抗原特异性杀伤活性、ELISpot试验和细胞内细胞因子染色。用这种方法研究抗肿瘤免疫的工作是通过将选定的T细胞克隆与抗原阴性的肿瘤细胞系共培养,这些肿瘤细胞系携带来自原始黑色素瘤细胞的基因组DNA或cDNA文库。这些研究鉴定了几种经典的黑色素瘤抗原,MAGE-A1、MAGE-A2、MAGE-A3和MART-1。同样的方法也被用来筛选非肥胖糖尿病小鼠中致病CD8+T细胞识别的自身抗原。另一种已知的肿瘤抗原NY-ESO-1是用一种名为重组cDNA表达文库(SEREX)(图2a)的血清学分析的方法鉴定的。多肽脉冲也被广泛用于富集抗原特异性T细胞。研究者使用一组平铺的18-mer NY-ESO-1多肽鉴定了四个NY-ESO-1特异性TCR。类似的方法也被用来富集病原体特异性T细胞。此外,单链三聚体(SCT)和合成mini基因也被广泛用于扩增或筛选抗原特异性CD8+T细胞(图2a)。Discovery of T cell antigens by high-throughput screening of synthetic minigene libraries使用合成mini基因筛选鉴定了来自1型糖尿病患者中自身反应性T细胞靶向的胰岛自身抗原的两个新表位。上述方法的主要障碍是需要大量表达TCR的T细胞。
一种常用的策略涉及使用pMHC多聚体,这是MHC分子的可溶性低聚形式。自从第一份报告显示pMHC多聚体可以与TCR提供足够的结合稳定性以来,荧光标记的pMHC多聚体与流式细胞术相结合,已被广泛用于体外可视化和分离抗原特异性T细胞(图2b)。然而,限制监测T细胞反应规模的一个主要限制因素是可以同时产生和使用的不同pMHC复合体的数量。为了克服这一挑战,已经建立了多肽交换技术(如UV交换)来促进各种pMHC多聚体的快速产生。pMHC多聚体方法的另一个主要缺点是,由于可用荧光染料的数量有限,在单个生物样本中只能检测到少量的抗原特异性。为了克服这一障碍,开发了两种并行的方法,使用荧光标记的多聚体的组合编码来同时检测数十个抗原特异性T细胞。第一种方法使用一组双色编码的pMHC多聚体,在单个样本中分析多达28个抗基因特异性。第二种多价码方法探索所有可能的荧光团组合,以检测和富集多达63种不同的T细胞特异性。使用这种方法,靶向两种不同新抗原的肿瘤浸润性淋巴细胞,并监测抗CTLA4治疗后新抗原特异性T细胞反应的变化。值得一提的是,这两种方法都要求pMHC多聚体中使用的表位具有最小的序列相似性,以避免多肽之间的交叉反应。
使用重金属离子作为标签的质谱细胞术技术的进步为提高T细胞表位筛选的能力提供了新的策略(图2c)。通过结合质谱细胞术和pMHC多聚体染色,开发了一种方法,允许在单个人类样本中同时识别多达109个pMHC特异性,并表征20-30个额外的表面和细胞内表型标记。这种方法后来被用来评估接受检查点阻断免疫治疗的荷瘤小鼠外周新抗原特异性CD8+T细胞的表型特征。尽管这些进展增强了定义免疫相互作用的能力,但单个TCR可以识别的大量可能的肽,以及TCR谱系的极端多样性,需要更高的通量。
DNA条码pMHC多聚体的使用扩展了这一工具包(图2d)。Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes让基因DNA条形码标记的pMHC多聚体在单个临床样本中并行检测1,000多个多肽特异性。通过利用这种DNA条形码技术,设计了一种“一锅法”策略,包括对给定目标多肽的每一个氨基酸进行顺序替换,以表征TCR的相对氨基酸偏好。这项技术还被用来比较发作性睡病患者和健康人中发作性睡病相关多肽特异性CD8+T细胞的频率。此外,DNA条形码技术的升级版本,称为四聚体相关TCR测序(TetTCR-seq),可以进一步在高通量下进一步将抗原特异性与单细胞中的TCR序列联系起来。研究人员通过体外转录和翻译制造了荧光链霉亲和素偶联的DNA条形码pMHC四聚体,并通过下一代测序破译了单个细胞的TCRα和TCRβ序列。DNA条形码标签的理论复杂性高达10^10,潜在地允许更高的通量。这些策略为了解T细胞的相互作用和选择“安全的”TCR用于临床开发提供了巨大的希望。然而,使用pMHC多聚体需要预先了解抗原靶标。此外,pMHC多聚体的其他几个潜在瓶颈,如多肽合成的通量有限、pMHC多聚体生产的低效率(如要求为每个HLA优化多聚体和低亲和力多肽的结合效率低下)、最大文库大小(由于蛋白质浓度高而可能导致pMHC多聚体聚集)、pMHC多聚体(特别是MHC II)的不稳定性和PCR扩增偏差,都需要得到解决,以充分利用其潜力。
微流体技术的快速发展为研究TCR-pMHC相互作用提供了新的工具。例如,Functional TCR T cell screening using single-cell droplet microfluidics建立了一种使用活化T细胞核因子(NFAT)-EGFP报告器在活细胞中监测TCR-pMHC识别动力学的方法。Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and Blood描述了一种使用纳米颗粒条形码核酸细胞分选(NACS)从黑色素瘤患者的肿瘤和血液中计数和分离新抗原特异性T细胞的灵敏方法。发展的微流控抗原-TCR接合测序(MATE-seq)技术允许高通量分离和单细胞TCR测序新抗原特异性T细胞。
这些抗原导向的策略,尽管它们有缺点,但提供了有用的工具来研究抗原特异的T细胞反应,并识别或分离推定的治疗性TCR用于临床。
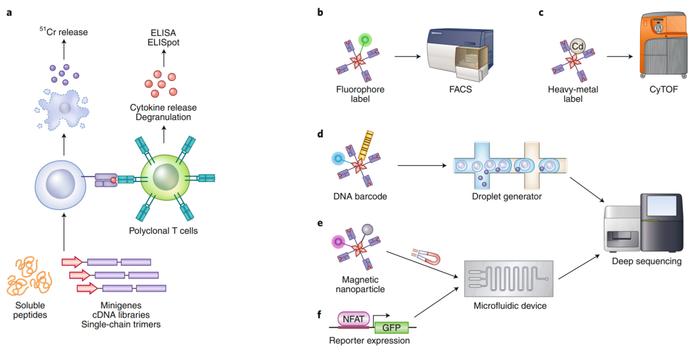
●图2 抗原导向方法。
以TCR为导向的方法
TCR导向方法的一般策略是从pMHC复合物的大型文库中筛选TCR或T细胞克隆(图3)。早期的TCR导向的方法使用组合多肽文库,将多肽中除一个位置以外的所有位置随机化,以筛选感兴趣的T细胞克隆(图3a)。例如,使用十肽文库鉴定H2-Ek-PCC(88-104)特异性T细胞克隆和HLA-DR2a-MBP(86-96)特异性T细胞克隆的所谓改变的多肽配体(APLs)。使用了200种OX9格式的多肽库混合物,其中O代表定义的氨基酸,X代表其余9个位置的20种天然氨基酸(半胱氨酸除外)中的任何一种,并评估了它们刺激T细胞克隆增殖的能力。基于导致最高增殖的多肽混合物,鉴定出一种APL(也称为模拟表位)。该方法也被用来确定MART-1表位的APL。
肽可以被遗传编码,并在细胞表面以杆状病毒、酵母或哺乳动物细胞库中的pMHC复合物的形式显示(图3b)。首先利用杆状病毒文库在Sf9昆虫细胞的表面展示流感血凝素(HA),然后产生可溶性的pMHC复合体和αβTCR。为了发现T细胞抗原,将pMHC复合体与杆状病毒蛋白gp64的跨膜区和胞浆区融合,将其锚定在Sf9细胞膜上。构建杆状病毒文库,其中确定的位置是随机的,得到的序列被用来感染Sf9细胞,然后用荧光标记的可溶性TCR进行染色,并通过FACS进行分选。经过三轮或四轮富集后,对单个病毒库进行测序,揭示TCRs识别的模拟表位。文库大小估计为0.3-1×10^5,仅为9-15mer文库理论极限的一小部分。杆状病毒展示的一个改进是使用酵母细胞在大型文库中展示pMHC复合体。感兴趣的蛋白质可以通过将其与α凝集素组分Aga2p融合在酵母细胞表面,Aga2p与细胞壁锚定的Aga1p相连。Aga1p-Aga2p连接已被用于展示单链可变片段的抗体、单链TCR和pMHC复合体。将单链pMHC复合体与Aga2p融合用于编码简并表位文库。用荧光标记的可溶性TCRs对酵母展示文库进行染色,用流式细胞仪进行分选,并对基因编码的pMHC进行测序。通过几轮富集,数十到数百个表位入围为潜在目标,然后使用位置权重矩阵制图,以识别自然出现的表位。这项技术已经用于小鼠和人的TCR识别MHC I和II。Deconstructing the peptide-MHC specificity of T cell recognition通过酵母展示I-Ek衍生的pMHC复合物,显示了显著的TCR与表位变体的交叉反应。Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes利用酵母展示技术,通过四轮筛选、位置权重矩阵和神经网络识别患者来源的结直肠癌私有TCRs的HLA-A0201限制性新表位靶点。
酵母展示的优势是从数十亿个pMHC复合体的真正简并文库开始,这不需要任何关于表位的先验知识。然而,它有一个主要的局限性,即需要修改MHC等位基因以增强适当的折叠、稳定性和pMHC呈现。相反,杆状病毒展示不需要这样的修饰。虽然酵母展示产生的一般都是模拟表位,但将它们映射到潜在的天然多肽的算法已经有了显著的改进。杆状病毒和酵母展示系统的一般限制是需要产生可溶的TCR,这是一个非稳健的过程。
与涉及酵母或杆状病毒展示的被动屏幕相比,哺乳动物细胞表面展示方法提供的屏幕中,同源pMHC复合体主动产生信号,用于将它们与无关的复合体区分开来(图3c)。基于哺乳动物细胞的抗原发现分析。第一种方法T cell antigen discovery via signaling and antigen-presenting bifunctional receptors使用称为信号和抗原提呈双功能受体(SABR)的受体,它由细胞外的pMHC复合体融合到细胞内的CD28-CD3ζ信号域组成。在TCR成功识别后,SABR向APC发起信号。使用NFAT-GFP Jurkat细胞,它在接收到CD28-CD3ζ信号时表达GFP,作为APC,可以识别特定TCR识别的SABR。在此基础上,构建了一个呈现HLA-A0201限制性表位的SABR文库,将其与表达TCR的Jurkat细胞孵育,对表达GFP的细胞进行分类,并对SABR基因进行测序以鉴定表位。成功地在一个约有12,000个表位的文库中识别出F5 TCR的同源表位。还演示了该技术在个体化新抗原发现中的应用。Deciphering CD4 T cell specificity using novel MHC-TCR chimeric receptors开发的一种类似的方法,将小鼠MHC II与TCRαβ的胞内和胞内结构域融合。这些受体被称为MCR-TCR,通过TCR复合体传递信号,其使用方式类似于SABRs。利用MCR-TCRs,确定了肿瘤特异性T细胞gp61表位和新抗原靶点的模拟表位。
在一个互补的方法中,这是两个相互作用的细胞之间的膜蛋白转移。表达TCR的Jurkat细胞与表达pMHC的K562细胞相互作用成功后,可将TCR从Jurkat细胞转移到K562细胞。在K562细胞上构建了单链HLA-A0201 pMHC复合体文库,并将其与表达TCR的Jurkat细胞孵育,对已摄取TCR的K562细胞进行了分选和测序,以确定它们所呈现的表位。T cell antigen discovery via trogocytosis利用这种方法,成功地鉴定了两个公共TCRs和一个新抗原反应性TCR的同源表位。还有一种类似的方法,称为T-Scan,使用工程颗粒酶B报告程序来标记表达表位编码mini基因的APC。T细胞识别APC后分泌的颗粒酶B导致APC中活性荧光蛋白的重建。可以使用T-scan直接筛选抗CMV特异性、病毒特异性或人类基因组衍生文库的原代T细胞。同时,使用由黄色荧光蛋白、青色荧光蛋白和含有颗粒酶B识别位点的连接肽接头组成的报告蛋白。当T细胞识别时,APC中的颗粒酶B对融合蛋白的切割导致黄色荧光蛋白产生的荧光共振能量转移(FRET)信号的丢失以及伴随而来的青色荧光蛋白信号的获得,从而使靶细胞得以分离。这些基于细胞的方法可以筛选表位,这一尺度介于抗原导向方法和酵母展示的尺度之间。这些方法不需要抗原的先验知识,但缩小可能的抗原空间会增强它们的结果。它们也可以与T细胞或TCR的寡克隆群体一起使用。然而,需要进一步的研究来确定所需的最低T细胞丰度,并在随后的检测中使用基于流体的方法或实验反卷积将T细胞与其同源APCs配对。
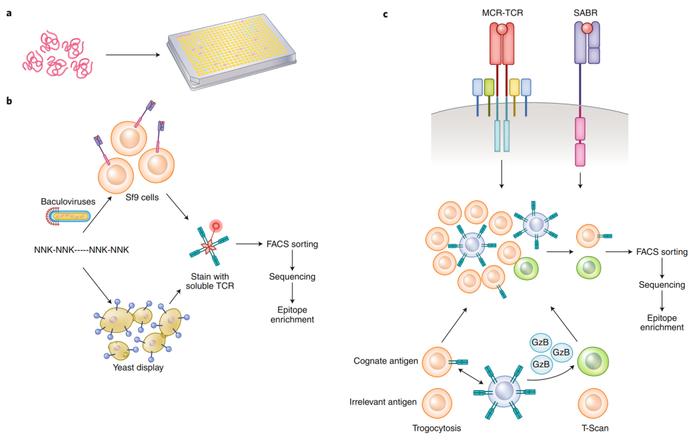
●图3 TCR导向方法。
计算方法
由于免疫系统中TCR的绝对数量,需要能够从α/β链序列中预测TCR的靶抗原的计算方法。高通量TCR测序的进展和TCR-pMHC特异性数据的积累导致了算法的出现,为计算抗原的发现铺平了道路。这些方法是建立在共享CDR3特征的TCR可能共享抗原特异性的概念上的。Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire对666个个体的8900万条TCRβ链进行了测序,并证明了预测CMV状态和HLA型的能力。同样,Antigen-specific TCR signatures of cytomegalovirus infection也是如此,表明特定的TCRβ家族与个体CMV的状态有关。
Identifying specificity groups in the T cell receptor repertoire研究表明在电子计算机中发现抗原是可以实现的。假设CDR3区域中的某些基序与某些抗原特异性有关。使用266436个独特的TCR序列,计算了所有可能的2-、3-和4-残基的“折叠富集度”和“富集率”。基于这些指标,抗原特异性谱系中的TCR序列落在一小部分高度相似的组中。开发了一种名为paratope热点分组淋巴细胞相互作用的算法(GLIPH),根据TCR可能的共享目标对其进行聚类。GLIPH考虑了TCR序列的相似性、结构信息、V基因使用和CDR3长度的偏差以及HLA类型。在验证实验中,GLIPH将结核支原体特异性TCR聚集在共享目标抗原的组中,并预测给定抗原的新TCR。在一项平行研究中Quantifiable predictive features define epitope-specific T cell receptor repertoires,分析了针对10个表位的4635个TCR,并观察到某些V基因片段和对的过度表达。使用了一种名为TCRdist的指标,该指标考虑了结构信息、氨基酸相似性和CDR长度来描述TCR之间的相似性。使用TCRdist以及TCR多样性度量TCRdiv和NN-dist,观察到TCR序列聚集成具有高度相似性的集群和不同的离群值。对流感特异性TCR进行了分类,并正确预测了它们的抗原。
这两项研究都表明,大多数识别相同抗原的TCR具有相同的序列和结构特征,并可根据这些特征将其分类为组。与这一过程相反,TCR可归因于可用于预测其抗原的簇。描述了考虑力场和结构信息或氨基酸的生化性质的类似方法。开发了被称为TCR实体肿瘤实用程序(TRUST)和使用T细胞受体排列的免疫相似性测量(iSMART)的算法,以识别与肿瘤相关的TCRs。GLIPH和TCRdist的一个关键区别是GLIPH只使用TCRβ序列,而TCRdist依赖TCRα和TCRβ链,这可能会导致更好的预测能力。虽然这些方法在预测TCR配体方面取得了重大进展,但它们仍有很长的路要走。一个主要问题是关于TCR-pMHC特异性的现有数据的规模,这可能通过广泛使用体外方法来解决。
选择抗原发现方法
对于研究人员来说,最重要的决定之一是为其特定需求选择适当的方法。在这里,根据几个问题提供关于选择哪种方法的指示,并描述基于对各种方法的性能的主观评估的实际考虑因素(图4和表1)。随着一些方法从概念验证转向更广泛的使用,这些考虑和建议将发生变化。
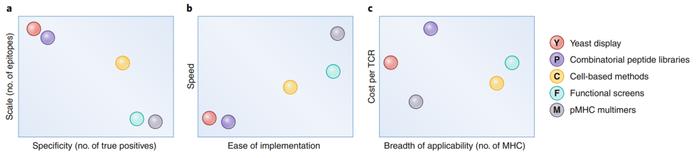
●图4 抗原发现方法的比较。a:规模与特异性。Scale表示一次可以筛选的pMHC数量;特异性是指识别的命中结果与实际表位的相似性。b:速度与实施的简易性。速度表示从头开始进行表位发现的速度;实施的简易性包括劳动强度和适应方法的便捷性。c:成本与适用范围。成本表示用于表位发现的每个TCR的成本;适用范围描述了一种方法用于不同MHC等位基因的容易程度。
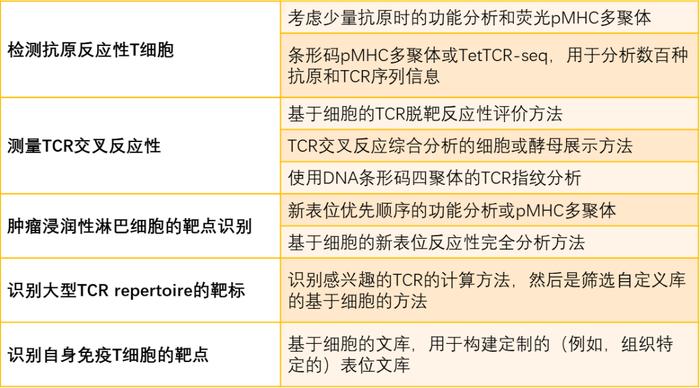
●表1 关于启动抗原发现项目的思考。
何去何从?
尽管最近取得了进展,但要实现对T细胞靶点的全面理解,仍需要做更多的工作。有几个挑战悬而未决:(1)抗原发现仅限于一次少量TCR;(2)MHC-II限制性TCR的配体发现尚未广泛存在;(3)在计算方法中,尚未达到临界质量的已知TCR-pMHC对来进行有意义的预测。然而,应该相信,TCR抗原发现技术的持续势头将解决其中的一些挑战。微流体技术的进步、更快和更高通量的深度测序和单细胞分析、更快的体外肽合成以及更好的大容量细胞培养和分选技术也将有助于扩大抗原发现的规模。其次,通过应用基于细胞的方法(如SABR和trogocytosis),能够发现基于MHC-II的表位,将使大量MHC II限制性TCR去孤儿化。预测表位与MHC II结合的工具的进展将有助于表位的发现。第三,越来越多记录在案的TCR-pMHC对将推动In Silico方法的迭代改进。TCR指纹和结构分析等补充方法也将有助于这些方法的发展。期望是在十年内,将能够完全根据TCRs的序列以高度精确度预测TCRs的靶抗原。这将是免疫学的一个范式转变,除了分析TCR谱系和基因表达外,考虑T细胞反应靶标的表位将成为常规。
