

转自:药时空
这里对新抗原的来源和生物学功能(上篇)、潜在的新抗原预测工具(中篇)以及基于新抗原的免疫治疗策略和临床应用(下篇)进行了全面的总结。此外,还讨论了基于新抗原的免疫疗法临床应用的机会和局限性,并提出了一些可能的解决方案。
新抗原的鉴定、预测和验证
从上篇众多来源中鉴定免疫原性新抗原是开发有效免疫疗法的关键一步。新抗原现在可以在整个癌症谱系中进行彻底筛选,这要归功于全外显子组测序(WES)、RNA-seq和来自TCGA的蛋白质组数据的融合。然而,考虑到肿瘤类型、肿瘤病变和患者的广泛差异,定制的免疫治疗需要基于不同的患者和肿瘤特征来检测和预测新抗原。为了预测免疫原性新抗原,需要鉴定基因组表达的突变以及MHC类型的细节,因为突变产生的肿瘤新抗原对免疫反应的顺序刺激依赖于几个变量,包括多肽的翻译和处理、MHC分子呈递突变多肽,以及pMHC复合体与TCR的亲和力。2种主要的识别新抗原表位的策略被开发出来:免疫基因组方法可以通过基于NGS的计算机方法创建虚拟多肽,免疫表位策略使用MS来分析MHC负载的多肽。最近开发了几种TCR引导的新抗原发现策略来系统地定位免疫原性新抗原。
01
体细胞突变的鉴定
通过使用NGS比较肿瘤和正常组织之间的基因变化,大大加快了免疫基因组学策略。目前,从NGS数据中检测可能的新抗原的过程的初始阶段是使用肿瘤和正常DNA的WES来绘制肿瘤特有的遗传异常。RNA-seq数据可以与WES相结合,以确定突变基因是否在肿瘤中表达。此外,在RNA-seq中还可以发现更多隐藏的生物信息,如有关拷贝数变化、微生物污染、转座元件、细胞类型和新抗原存在的信息。RNA-seq还可用于检测选择性剪接事件和估计突变等位基因表达的相对频率。通过使用可能检测染色体重排的配对测序等方法,基于NGS的TMB方法的预测值可能会大大提高。最近的研究表明,当假定NMD存在时,抗原肽是由具有移码突变和非典型剪接模式的转录本产生的。需要从全长转录本结构中获得准确的多肽序列,以便完全确定移码突变和异常异构体所产生的新抗原。使用纳米孔型测序仪MinION,全长转录组测序可能在适当的测序深度覆盖整个转录本,准确率约为90%,为目前的RNA-seq提供补充信息,以鉴定等位基因特异的转录和剪接。
基于癌症基因组数据,免疫基因组学技术预测了数百万可能的突变衍生的新抗原,但其中绝大多数并未在HLA结合肽的蛋白质组学谱中表现出来。免疫抗原学技术使高通量鉴定MHC结合多肽成为可能,它使用MS直接检测免疫沉淀和提取的MHC结合多肽。MS在验证计算机预测的新抗原方面取得了进展。将样品的串联质谱图与合成肽的串联质谱图进行比较,可以验证免疫基因组学方法预测的新抗原。特别是对于稀有的HLA同种异型和HLA-II配体,绘制肿瘤HLA配体图谱有助于在临床试验中发现新抗原特异性癌症免疫疗法的靶点。除了验证异常DNA序列或RNA表达产生的新抗原外,基于MS的蛋白质组学还为在蛋白质水平上检测新抗原提供了DNA和RNA研究所无法发现的“金标准”。例如,MS可用来检测由PTM产生的新的MHC相关的新抗原,这些新的MHC相关的新抗原在细胞转化过程中是不受调控的。此外,MS还与NGS整合,以进一步检测由体细胞突变、非编码RNA和蛋白酶体剪接产生的肿瘤特异性新抗原,这些都是基于全外显子组或转录组的测序技术所省略的。为了在蛋白质水平上更深入地了解新抗原,应该创建更方便和实用的工具,整合基因组、转录和蛋白质组数据用于基于免疫表位的新抗原检测。
02
计算机预测新抗原
基于NGS数据,已经创建了虚拟肽库,并通过计算机方法发现了潜在的新抗原。简单地说,新抗原预测的典型工作流程可概括为以下步骤:(i)突变鉴定,(ii)HLA分型,(iii)基于HLA结合亲和力的新抗原筛选和优先排序,以及(iv)基于T细胞分析的免疫原性新抗原的实验验证(图3)。
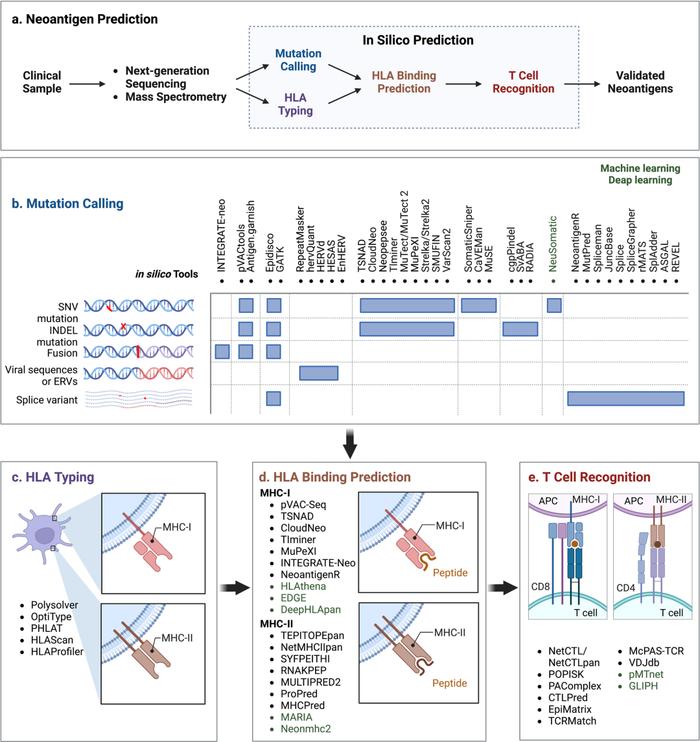
●图3 新抗原预测的计算工作流程。目前可用于从体细胞突变预测新抗原的生物信息学管道共享四个主要计算模块:(i)来自肿瘤WGS、WES数据和RNA-Seq的HLA分型;(ii)使用一组体细胞突变和剪接变体调用突变多肽;(iii)HLA结合预测;以及(iv)T细胞识别预测。用于突变调用的计算机工具如下所示。突变调用:INTEGRATE-neo,neoFusion,pVACtools, Epidiso,GATK and Antigen.garnish,Spliceman,MutPred,REVEL,rMATS,pVACseq,Neopepsee, MuPeXI,RepeatMasker,CloudNeo,Tlminer,MuTect/MuTect2,Strelka/Strelka2, SMUFIN,VarScan2,SomaticSniper,CaVEMan, MuSE, cgpPindel, SvABA, RADIA, NeuSomatic, NeoantigenR, MutPred, JuncBase, Splice, SpliceGrapher, rMATS, SplAdder, ASGAL, REVEL, TSNAD, HERVd, HESAS and EnHERV, hervQuant。HLA分型:Polysolver, OptiType, HLAreporter, PHLAT, HLAScan, HLAProfiler。HLA结合亲和力:NetMHCpan, NetMHCIIpan4.0, MixMHC2pred, MARIA, neomhc2, pVAC-Seq, TIminer, HLAthena, DeepHLApan, TEPITOPEpan, NetMHCIIpan, SYFPEITHI, RNAKPEP, MULTIPRED2, ProPred, MHCPred, MARIA, Neonmhc2, EDGE。T细胞识别:NetCTL/NetCTLpan, POPISK, PAComplex, CTLPred, EpiMatrix, TCRMatch
03
HLA分型
新抗原通常由CD8+T细胞的MHC-I和CD4+T细胞的MHC-II以细胞特异性的方式递送,与其他抗原非常相似。人类有24,000多个不同的HLA-I(HLA-A、-B和-C)和HLA-II(HLA-DR、HLA-DQ和HLA-DP)等位基因,它们的混合导致多态多样性。患者的HLA等位基因决定了他们的肿瘤特异性新抗原库,这些新抗原库将提供给T细胞识别。此外,在40%的非小细胞肺癌中发生的HLA-LOH会损害新抗原的呈递,从而促进免疫逃避。因此,在新抗原预测中最重要的初始步骤之一是确定患者的HLA基因型。几种计算方法现在可应用于NGS数据来实现这一目标。大多数方法依赖于从WES或WGS获得的DNA衍生的NGS数据。例如,Optiype和Polysolver是用于鉴定I类HLA等位基因的优秀工具。开发了一种生物信息学工具LOHHLA,用于准确测量等位基因特异性的HLA拷贝数。这些工具,包括HISAT genotype,ATHLATES和HLA-HD,既可用于I类分型,也可用于II类分型。RNA-seq数据也有可用工具,如arcasHLA,seq2HLA和HLAProfiler,利用平等地涵盖两个完全表达的亲本等位基因的无偏数据集的优势来分型HL A等位基因。新开发的基于RNA-Seq数据的方法为HLA分型和生物标记物研究带来了一个新的维度,尽管Optitype发现WES在HLA分型方面比RNA-Seq数据产生了更好的结果。
04
突变和变异调用
通过比较同一患者肿瘤和正常组织的NGS数据,可以预测由体细胞突变引起的突变多肽。WES是新抗原预测的NGS数据的推荐来源,因为它通过关注基因组的蛋白质编码区提供了最高的突变覆盖率。计算分析包括数据的预处理和质量控制、体细胞突变的变异,以及利用公共基因组、转录组和蛋白质组序列数据库预测改变的蛋白质和功能影响。根据用于筛选假定新抗原的战略,这些技术可分为两类:基于逐步分析的过滤策略和基于综合评分系统的过滤策略。高效的一站式工具接受WES/WGS和RNA-seq数据作为输入,并基于选定的截止指标执行一系列筛选步骤,例如多肽与MHC分子的结合亲和力、序列覆盖率、变异等位基因频率和基因表达,以消除假阳性并生成潜在新抗原的列表。基于综合评分系统的过滤技术通过基于显著的新肽特征的定量评分来评估新抗原的免疫原性,包括突变肽和正常肽的等级亲和力、突变等位基因的频率和基因表达的量,以实验地评估发现的新肽的免疫原性。最近,还提出了一种基于机器学习模型的免疫原性评估评分方法,优化了新抗原的准确预测,减少了假阳性。
05
HLA结合和新抗原呈递的预测
已经创建了许多计算机预测工具,用于基于MHC分子加工和呈现的新抗原的计算机发现,包括NetChop、NetCTL和NetCTLpan (图3)。通过将HLA配体组数据结合到机器学习算法中,如线性回归和人工神经网络,积极提高预测能力。体外肽-HLA结合数据集被用于训练机器学习模型,NetMHCpan和MHCflurry是当前HLA配体识别线的主要组件。值得注意的是,与先进的方法相比,NetMHCpan通过结合结合亲和力数据和MS多肽组数据的信息来提高肿瘤新抗原的预测性能,以给出针对MHC-I的“泛特异性”机器学习策略。最近的两项研究创建了被称为MSIntrinsic和EDGE的计算框架,它们在使用从RNA-Seq和 LC-MS/MS数据中获得的HLA多肽预测HLA抗原方面非常有效。基于LC-MS/MS收集的24,000个HLA-I多肽,神经网络预测算法MSIntrinsic的阳性预测值(PPV)平均比以前基于亲和力的预测值(PPV)高出30%。类似的发现由EDGE得出,发现采用深度学习结构来利用蛋白质组和转录组数据鉴定HLA配体可以将HLA抗原预测的准确性提高9倍。
越来越多的证据表明,MHC-II新抗原在抗肿瘤免疫反应中具有重要意义。利用人工神经网络已经开发了一系列用于预测MHC-II结合表位的计算技术,包括NetMHCII、NetMHCIIpan、SYFPEITHI、RNAKPEP、MULTIPRED2、ProPred和MHCPred。然而,与MHC-I分子相比,目前对MHC-II多肽结合亲和力的计算预测不那么精确。首先,与MHC-I分子相比,MHC-II结合肽在肽长度和结合序列基序方面更加混杂。其次,MHC-II分子中α和β链的多态性也极大地扩展了多肽结合特异性的多样性。最近,基于转录组和MS数据的计算方法已经发展起来。由MARIA训练的深度学习模型,结合了测序数据和天然存在的MHC-II配体,当与已知的MHC-II配体交叉验证时,被证明优于淋巴瘤数据集中最广泛使用的预测因子NetMHCIIpan3.1。然而,为了证明它的稳健性和有效性,需要使用大量的数据集进行更多的研究。
鉴于多个过程控制新抗原提呈,可以推断,仅提高结合亲和力并不能准确反映细胞处理和CD8+T细胞反应。其他特性,包括蛋白酶体切割、多肽进入内质网的运输和HLA等位基因,与多肽和MHC分子之间的结合亲和力相结合,以优先考虑可能的新抗原。
06
候选新抗原免疫原性的评估与验证
众所周知,一个免疫原性新抗原必须满足两个或多个要求,主要的瓶颈是合适的MHC分子递呈和有效的TCR识别。根据最近的研究,大多数通过MHC分子递呈预测的新抗原不会引发免疫反应。因此,在评估潜在新抗原的免疫原性时,考虑pMHC复合体的TCR识别是至关重要的。计算机技术中有许多方法可以预测新抗原特异性T细胞识别。最常用的方法是NetCTL/NetCTLspan,它通过结合MHC结合、C端切割亲和力和TAP转运蛋白来生成综合评分,而不是直接预测T细胞结合。最近的研究使用机器学习或深度学习技术来预测TCR-肽/-pMHC结合。在几个手动策划的数据库(包括 McPAS-TCR 和 VDJdb)中的一批 TCR 库注释允许训练 TCR 特异性预测因子并与感兴趣的 TCR 进行匹配。McPAS-TCR提供了与各种病理相关联的TCR序列的列表,而VDJdb提供了TCR的详细描述:基于以表位为中心的方法对TCR的相互作用进行注释,而不是基于潜在的生物学背景。除了鉴定TCR-pMHC配对之外,像pMTnet和GLIPH这样的聚类法也可以对识别相同表位的TCR进行聚类并预测它们HLA的限制。由于TCR与pMHC配体的亲和力较低,预测TCR和pMHC在计算机中的结合亲和力仍然是具有挑战性的。
为了更准确地评估新抗原在免疫治疗中的可能应用,对其T细胞反应性的实验验证是至关重要的。新抗原反应性T细胞已经通过基于T细胞的检测、多色标记的MHC四聚体、酶联免疫吸附斑点试验(ELISpot)和T细胞库分析进行验证或筛选。T细胞免疫原性测定是评估候选新抗原免疫原性的最直接方法。癌症外显子组/RNA-seq发现的一整套可能的突变多肽可以用来自癌症患者或健康捐赠者的T细胞进行测试。在多肽刺激后,通过流式细胞仪测量T细胞激活标志物4-1BB和OX-40以及ELISpot分析干扰素的产生,来测量体外扩增的新抗原特异性T细胞的反应性。多色标记的MHC四聚体允许使用DNA条形码、稀土元素编码或多肽的荧光编码来高度敏感且不需要太多材料来评估T细胞针对各种潜在表位的反应性。这些技术依赖于表位预测,而且通量很低,因为它们只能有效地产生人MHC I类等位基因的子集。将单细胞RNA测序(scRNA-seq)与响应细胞群的TCR测序相结合可用于提高检测的灵敏度。scRNA-seq用于发现与串联微基因(TMG)转染或肽刺激的抗原呈递细胞(APC)共培养的TIL中表达高水平IFN-γ和IL-2的细胞相连的配对TCR序列。基于WES引导的新抗原预测和短期多肽刺激的T细胞培养的TCR测序,突变相关的新抗原功能扩增特异性T细胞(MANAFEST)检测敏感地表征了新抗原特异性的TCRVβ克隆型。MANAFEST检测兼容所有的HLA单倍型,可以追踪福尔马林固定石蜡包埋(FFPE)和/或冰冻组织中的新抗原特异性T细胞。除了评估TCRVβ克隆型的肿瘤特异性外,MANAFEST还可以研究新抗原特异性T细胞反应随时间的动态变化,并使用治疗前或治疗后获得的液体活检监测免疫治疗的效果。
几种无偏倚的TCR引导的新抗原发现策略已经被开发出来,以系统地分析新抗原特异性TCR。酵母展示的pMHC文库可用于发现新抗原特异性TCR。然而,制备可溶性TCR试剂是一个耗时的过程。如果没有新抗原的内源性加工或T细胞的功能激活,所识别的随机多肽可能不代表生理上的TCR-pMHC相互作用。为了克服这些缺点,两种创新的策略利用不同的生物学过程来标记共培养系统中的靶细胞。一种方法是利用被称为信号和抗原提呈双功能受体(SABRs)的嵌合受体,它可以在pMHC-TCR相互作用后诱导TCR样信号。SABRs能够成功地鉴定TCR-pMHC相互作用,这既可用于已知的公共TCR,也可用于私有的新抗原特异性TCR。胞啃是一种膜转移过程,由基于细胞的选择平台用于TCR配体的发现。TCR-pMHC的相互作用导致同源靶细胞的特异性标记,然后对其进行分离和测序以识别新抗原特异性的TCR。此外,推测的pMHC在BATTLES中显示在光谱编码磁珠上,便于研究生理力量下的新抗原特异性T细胞反应。T-Scan是一种独立于预测算法的TCR表位扫描方法,它依赖于T细胞杀伤的生理活性,而不仅仅是评估TCR-pMHC的结合亲和力,使询问的抗原空间比以前的方法大得多。因此,这些新出现的发现TCR配体的方法将有助于研究候选新抗原的免疫原性,为免疫治疗提供新的靶点。
参考文献:
Okada, M., et al. Identification of neoantigens in cancer cells as targets for immunotherapy. Int. J. Mol. Sci. 23, 2594 (2022).
Finotello, F., et al. Next-generation computational tools for interrogating cancer immunity. Nat. Rev. Genet. 20, 724–746 (2019).
Xie, N., Shen, G., Gao, W. et al. Neoantigens: promising targets for cancer therapy. Sig Transduct Target Ther 8, 9 (2023)
Jurtz, V. et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
